Chorea Huntington Wann Bricht Es Aus

Huntington-Krankheit, auch Chorea Huntington genannt, ist eine verheerende neurodegenerative Erkrankung, die das Leben der Betroffenen und ihrer Familien dramatisch verändert. Die Frage, wann diese Krankheit ausbricht, ist oft die erste und dringendste Frage, die sich Menschen stellen, die ein Risiko tragen. Sie ist mit Unsicherheit, Angst und der Notwendigkeit verbunden, sich auf eine Zukunft vorzubereiten, die möglicherweise ganz anders aussieht als erwartet. Wir verstehen diese Herausforderungen und möchten Ihnen helfen, die komplexe Natur des Krankheitsbeginns bei Chorea Huntington zu verstehen.
Was ist Chorea Huntington?
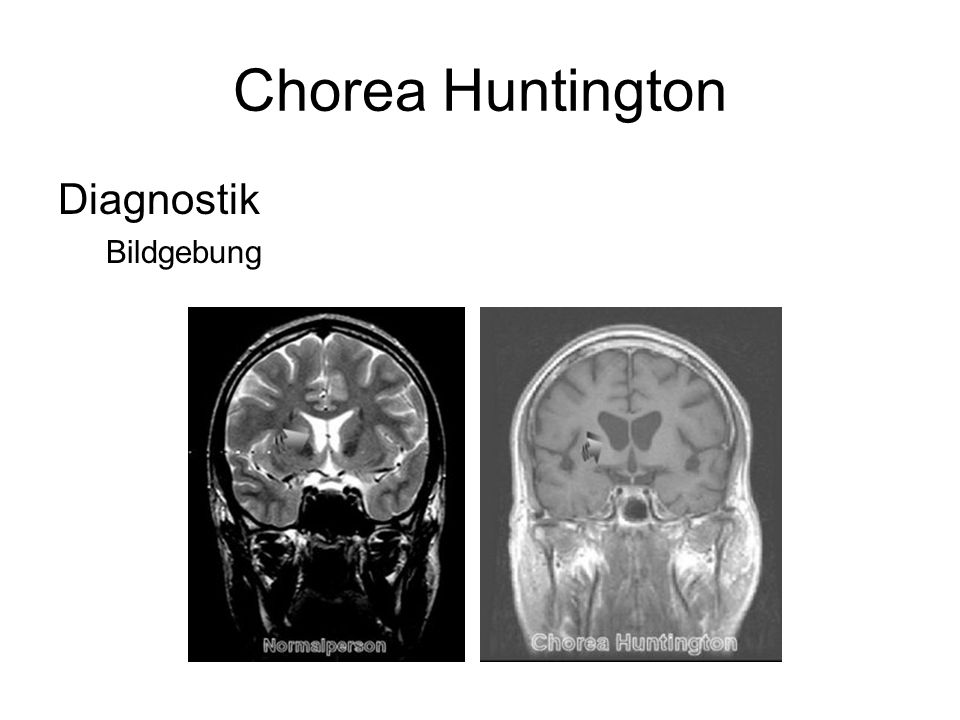
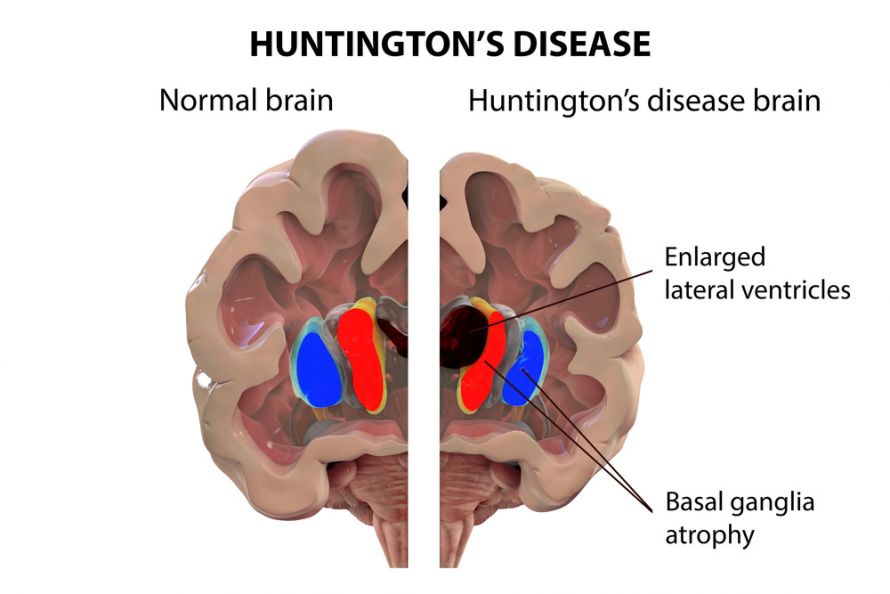
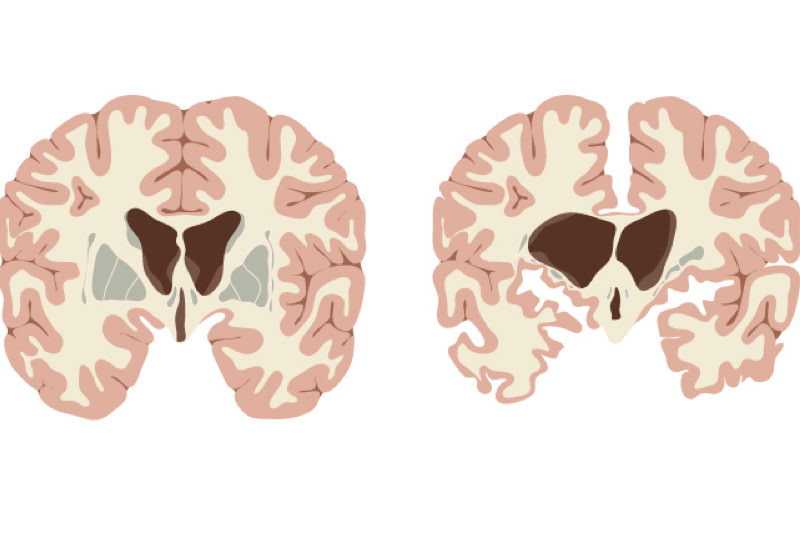
Chorea Huntington ist eine erbliche Erkrankung, die durch den fortschreitenden Abbau von Nervenzellen im Gehirn gekennzeichnet ist. Dies führt zu einer Vielzahl von Symptomen, die sich auf Bewegung, Kognition und psychische Gesundheit auswirken. Die Erkrankung wird autosomal dominant vererbt, was bedeutet, dass eine einzige Kopie des mutierten Gens ausreicht, um die Krankheit auszulösen. Das heißt, wenn ein Elternteil die Krankheit hat, besteht für jedes Kind eine Wahrscheinlichkeit von 50%, die Krankheit ebenfalls zu erben.
Die genetische Grundlage
Die Ursache für Chorea Huntington ist eine Mutation im Huntingtin-Gen (HTT), das sich auf Chromosom 4 befindet. Diese Mutation beinhaltet eine Verlängerung einer CAG-Wiederholungssequenz innerhalb des Gens. Eine CAG-Wiederholung ist eine Abfolge der Basen Cytosin (C), Adenin (A) und Guanin (G) im DNA-Code. Menschen ohne Huntington haben in der Regel weniger als 36 CAG-Wiederholungen. Bei Menschen mit Huntington-Krankheit sind es 40 oder mehr. Je höher die Anzahl der Wiederholungen, desto früher kann die Krankheit ausbrechen. Bereiche zwischen 36 und 39 Wiederholungen werden als "reduzierte Penetranz" bezeichnet. Personen mit dieser Anzahl von Wiederholungen entwickeln möglicherweise die Krankheit im Laufe ihres Lebens nicht oder erst im hohen Alter.
Wann bricht Chorea Huntington aus?
Das Alter des Krankheitsbeginns bei Chorea Huntington ist sehr variabel und kann zwischen dem Kindesalter und dem hohen Alter liegen. Allerdings beginnt die Erkrankung typischerweise zwischen dem 30. und 50. Lebensjahr.
- Früher Krankheitsbeginn: Selten beginnt die Erkrankung vor dem 20. Lebensjahr. Dies wird als juvenile Huntington-Krankheit bezeichnet und ist oft mit einer größeren Anzahl von CAG-Wiederholungen verbunden. Die Symptome können sich von denen des typischen Krankheitsbeginns unterscheiden und umfassen häufig Steifheit, Anfälle und Lernschwierigkeiten.
- Typischer Krankheitsbeginn: Der häufigste Beginn liegt zwischen dem 30. und 50. Lebensjahr. Die Symptome entwickeln sich in der Regel schleichend und können zunächst subtil sein.
- Später Krankheitsbeginn: In einigen Fällen kann die Krankheit erst nach dem 60. Lebensjahr ausbrechen. Der spätere Beginn kann schwieriger zu diagnostizieren sein, da die Symptome möglicherweise mit anderen altersbedingten Erkrankungen verwechselt werden.
Faktoren, die den Krankheitsbeginn beeinflussen
Neben der Anzahl der CAG-Wiederholungen können auch andere Faktoren den Zeitpunkt des Krankheitsbeginns beeinflussen. Dazu gehören:
- Genetische Modifikatoren: Es gibt Hinweise darauf, dass andere Gene neben HTT den Krankheitsbeginn beeinflussen können. Diese Gene könnten die Art und Weise beeinflussen, wie das mutierte Huntingtin-Protein die Nervenzellen schädigt.
- Umweltfaktoren: Obwohl die Genetik eine entscheidende Rolle spielt, könnten Umweltfaktoren wie Ernährung, Exposition gegenüber Toxinen und Lebensstil ebenfalls eine Rolle spielen. Die Forschung in diesem Bereich ist jedoch noch im Gange.
- Epigenetische Faktoren: Epigenetik beschreibt Veränderungen der Genexpression, die nicht durch Veränderungen der DNA-Sequenz selbst verursacht werden. Diese Faktoren können beeinflussen, wie das Huntingtin-Gen exprimiert wird und somit den Krankheitsbeginn beeinflussen.
Symptome von Chorea Huntington
Die Symptome von Chorea Huntington sind vielfältig und können sich von Person zu Person stark unterscheiden. Sie werden in drei Hauptkategorien unterteilt:
- Bewegungsstörungen: Chorea, unwillkürliche, ruckartige Bewegungen, ist ein Kennzeichen der Erkrankung. Andere Bewegungsstörungen können Dystonie (anhaltende Muskelkontraktionen), Rigidität und Bradykinesie (Verlangsamung der Bewegungen) sein.
- Kognitive Beeinträchtigungen: Kognitive Probleme umfassen Schwierigkeiten bei der Planung, Organisation, Entscheidungsfindung und dem Gedächtnis. Auch die exekutiven Funktionen, die für zielgerichtetes Verhalten wichtig sind, sind oft beeinträchtigt.
- Psychische Störungen: Depressionen, Angstzustände, Reizbarkeit, Zwangsstörungen und Psychosen können auftreten. Diese psychischen Symptome können erhebliche Auswirkungen auf die Lebensqualität haben.
Der Einfluss auf das Leben
Die Symptome der Huntington-Krankheit wirken sich auf nahezu jeden Aspekt des Lebens der Betroffenen aus. Bewegungsstörungen können es schwierig machen, alltägliche Aufgaben wie Essen, Anziehen und Gehen zu erledigen. Kognitive Beeinträchtigungen können die Fähigkeit zur Arbeit, zur Verwaltung von Finanzen und zur Teilnahme an sozialen Aktivitäten beeinträchtigen. Psychische Probleme können zu sozialer Isolation, Beziehungsstress und einem erhöhten Suizidrisiko führen.
Es ist wichtig, sich daran zu erinnern, dass jeder Mensch die Huntington-Krankheit anders erlebt. Es gibt keine "typische" Verlaufsform und die Symptome können sich im Laufe der Zeit verändern.
Diagnose und genetische Beratung
Die Diagnose von Chorea Huntington basiert in der Regel auf einer Kombination aus neurologischer Untersuchung, Familienanamnese und genetischer Testung. Eine genetische Testung kann durchgeführt werden, um die Anzahl der CAG-Wiederholungen im Huntingtin-Gen zu bestimmen.
Gentests sind eine persönliche Entscheidung. Personen mit einer Familienanamnese von Chorea Huntington, die keine Symptome zeigen, können sich einem prädiktiven Gentest unterziehen, um festzustellen, ob sie das mutierte Gen geerbt haben. Dieser Test kann jedoch erhebliche emotionale und psychologische Auswirkungen haben. Es ist ratsam, sich vor und nach der Testung genetisch beraten zu lassen, um die Vor- und Nachteile sowie die ethischen Implikationen zu verstehen.
Pränatale Tests sind ebenfalls möglich, wenn ein Elternteil die Krankheit hat. Dies wirft jedoch komplexe ethische Fragen auf, die sorgfältig abgewogen werden müssen.
Behandlung und Management
Derzeit gibt es keine Heilung für Chorea Huntington. Die Behandlung konzentriert sich auf die Linderung der Symptome und die Verbesserung der Lebensqualität.
- Medikamente: Medikamente können zur Behandlung von Bewegungsstörungen, Depressionen, Angstzuständen und anderen psychischen Symptomen eingesetzt werden. Beispiele hierfür sind Tetrabenazin oder Deutetrabenazin zur Behandlung von Chorea und Antidepressiva zur Behandlung von Depressionen.
- Physiotherapie: Physiotherapie kann helfen, die Kraft, Flexibilität und das Gleichgewicht zu verbessern.
- Ergotherapie: Ergotherapie kann helfen, Strategien zur Bewältigung alltäglicher Aufgaben zu entwickeln und die Selbstständigkeit zu fördern.
- Sprachtherapie: Sprachtherapie kann helfen, Sprach- und Schluckbeschwerden zu behandeln.
- Psychotherapie: Psychotherapie kann helfen, mit den emotionalen und psychologischen Herausforderungen der Huntington-Krankheit umzugehen.
- Unterstützende Pflege: Unterstützende Pflege umfasst Ernährungsberatung, soziale Unterstützung und Palliativpflege.
Die Forschung schreitet stetig voran. Es gibt eine Reihe vielversprechender Forschungsansätze, darunter Gentherapie, Medikamente zur Reduzierung des Huntingtin-Proteins und Therapien zum Schutz von Nervenzellen.
Umgang mit der Ungewissheit
Das Leben mit dem Risiko, an Chorea Huntington zu erkranken, ist mit grosser Ungewissheit verbunden. Es ist wichtig, sich daran zu erinnern, dass es Ressourcen und Unterstützung gibt, die Ihnen helfen können, mit diesen Herausforderungen umzugehen.
- Selbsthilfegruppen: Der Austausch mit anderen, die ähnliche Erfahrungen machen, kann unglaublich hilfreich sein.
- Beratung: Ein Therapeut kann Ihnen helfen, mit Angst, Depressionen und anderen emotionalen Problemen umzugehen.
- Planung: Die Planung für die Zukunft, einschliesslich finanzieller Planung, rechtlicher Dokumente und Pflegeplanung, kann Ihnen ein Gefühl der Kontrolle geben.
- Auf sich selbst aufpassen: Achten Sie auf Ihre körperliche und geistige Gesundheit. Essen Sie gesund, treiben Sie regelmässig Sport und nehmen Sie sich Zeit für Aktivitäten, die Ihnen Freude bereiten.
Es ist wichtig, sich daran zu erinnern, dass eine genetische Veranlagung für Chorea Huntington nicht bedeutet, dass Sie die Krankheit unbedingt entwickeln werden oder dass Ihr Leben vorbei ist. Viele Menschen mit dem Gen leben ein erfülltes und sinnvolles Leben.
Die Frage "Wann bricht Chorea Huntington aus?" hat keine einfache Antwort. Der Krankheitsbeginn ist variabel und wird von einer Vielzahl von Faktoren beeinflusst. Das Wichtigste ist, sich zu informieren, sich mit Ressourcen zu vernetzen und proaktiv Schritte zu unternehmen, um Ihre Gesundheit und Ihr Wohlbefinden zu erhalten. Sie sind nicht allein.
Welche Fragen haben Sie noch, und welche Schritte möchten Sie als Nächstes unternehmen, um sich über Chorea Huntington zu informieren und Unterstützung zu suchen?